Tuning the Electronic and Magnetic Properties of Double Transition Metal (MCrCT2, M = Ti, Mo) Janus MXenes for Enhanced Spintronics and Nanoelectronics
Swetarekha Ram, Namitha Anna Koshi, Seung-Cheol Lee, and Satadeep Bhattacharjee
The Journal of Physical Chemistry C
Abstract
Tuning the Electronic and Magnetic Properties of Double Transition Metal (MCrCT2, M = Ti, Mo) Janus MXenes for Enhanced Spintronics and Nanoelectronics
Janus MXenes, a new category of two-dimensional (2D) materials, show promising potential for advances in optoelectronics, spintronics, and nanoelectronics. Our theoretical investigations not only provide interesting insights but also highlight the promise of Janus MCrCT2 (M = Ti, Mo; T = O, F, OH) MXenes for future spintronic applications and highlight the need for their synthesis. Electronic structure analysis shows different metallic and semimetallic properties: MoCrCF2 exhibits metallic property, TiCrC(OH)2 and MoCrCO2 exhibit near semimetallicity with spin polarization values of 61 and 86%, respectively, while TiCrCO2 and TiCrCF2 are completely half-metallic with 100% spin polarization at the Fermi level. All studied Janus MXenes exhibit intrinsic ferromagnetism, which is mainly attributed to the chromium (Cr) atoms, as shown by the spin density difference plots. Among them, the TiCrCO2 monolayer stands out with the highest exchange constant and ferromagnetic transition temperature (Tc). Notably, the O-terminated Janus MXenes exhibit weak perpendicular magnetic anisotropy, in contrast to the in-plane anisotropy observed for F and OH-terminated MXenes, making them particularly interesting for future spintronic applications, which we further demonstrate with micromagnetic simulation which reveal distinct current-induced switching behaviors in these Janus MXenes with different surface terminations.
94
Controlling Moisture for Enhanced Ozone Decomposition: A Study of Water Effects on CeO2 Surfaces and Catalytic Activity
Controlling Moisture for Enhanced Ozone Decomposition: A Study of Water Effects on CeO2 Surfaces and Catalytic Activity
This study investigates the catalytic degradation of ground-level ozone on low-index stoichiometric and reduced CeO2 surfaces using first-principles calculations. The presence of oxygen vacancies on the surface enhances the interaction between ozone and the catalyst by serving as active sites for adsorption and decomposition. Our results suggest that the {111} surface has superior ozone decomposition performance due to unstable oxygen species resulting from reactions with catalysts. However, when water is present, it competes with ozone molecules for these active sites, resulting a reduced catalytic activity or water poisoning. A possible solution could be heat treatment that reduces the vacancy concentration, thereby increasing the available adsorption sites for ozone molecules while minimizing competitive adsorption by water molecules. These results suggest that controlling moisture content during operation is crucial for the efficient use of CeO2-based catalysts in industrial applications to reduce ground-level ozone pollution.
93
Tuning of nodal line states via chemical alloying in Co2CrX( X=Ga, Ge) Heusler compounds for a large anomalous Hall effect
Ujjawal Modanwal, Gaurav K. Shukla, Ajit K. Jena, Satadeep Bhattacharjee, Sunil Wilfred D'Souza, Jan Minár, and Sanjay Singh
Physical Review Materials
Abstract
Tuning of nodal line states via chemical alloying in Co2CrX( X=Ga, Ge) Heusler compounds for a large anomalous Hall effect
Topological materials have attracted significant interest in condensed matter physics for their unique topological properties leading to potential technological applications. Topological nodal line semimetals, a subclass of topological materials, exhibit symmetry-protected nodal lines, where band crossings occur along closed curves in the three-dimensional Brillouin zone. When the nodal lines are gapped out due to perturbation in the Hamiltonian, a large Berry curvature (BC) arises in the surrounding area of the gapped nodal line, leading to exotic anomalous transport responses. In this paper, we studied the Co2CrX (X=Ga, Ge) Heusler compounds that exhibit mirror symmetry-protected nodal line states below the Fermi level. The BC calculation yields anomalous Hall conductivity (AHC) of about 292 and 217 S/cm for Co2CrX (X=Ga, Ge), respectively, at the Fermi level, which increases by up to 400% at the nodal line energy level. We theoretically analyzed that 20% and 60% zinc (Zn) alloying in Co2CrX (X=Ga, Ge) effectively lowers the Fermi level by 50 meV and 330 meV, respectively, aligning with the protected crossings. Consequently, we identified Co2CrGe0.4Zn0.6 and Co2CrGa0.8Zn0.2 as compositions to achieve the significant AHC of 800 and 1300 S/cm, respectively. The explicit AHC calculation for these alloyed compositions is in good agreement with our predictions. Our findings highlight that chemical alloying is an efficient way to enhance AHC in nodal line hosting materials.
92
Improved resistance to water poisoning of Pd/CeO2 monolithic catalysts by heat treatment for ozone decomposition
Improved resistance to water poisoning of Pd/CeO2 monolithic catalysts by heat treatment for ozone decomposition
Durability is a crucial requirement in heterogeneous catalysis; however, many catalysts suffer from severe deactivation in humid conditions due to water poisoning. Ozone, as a significant air pollutant, should be efficiently removed through catalytic decomposition, making it imperative to develop a water-tolerant monolithic catalyst for practical air purification. In this study, we present highly durable Pd/CeO2 monolithic catalysts resistant to water poisoning achieved through a simple heat treatment of the ceria support. The heat treatment controlled the ceria surface properties, including oxygen vacancy defects, surface oxygen, and basicity, thereby improving resistance to water poisoning. When Pd/CeO2 monolithic catalysts were used in bench-scale ozone decomposition under humid conditions, the catalyst heat-treated at 900 °C exhibited superior performance without experiencing deactivation due to water poisoning. Modulating the ceria surface properties plays a pivotal role in enhancing water resistance, and heat-treated Pd/CeO2 monolithic catalysts stand as a promising candidate for practical ozone decomposition in air purification applications.
91
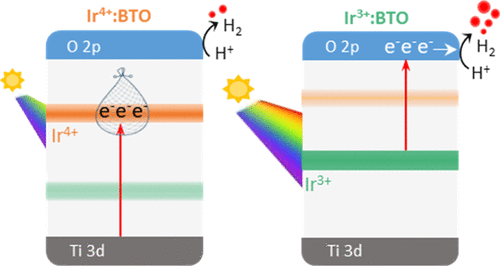
Unveiling Valence State-Dependent Photocatalytic Water Splitting Activity and Photocathodic Behavior in Visible Light-Active Iridium-Doped BaTiO3
Sujana Chandrappa, Stephen Nagaraju Myakala, Namitha Anna Koshi, Simon Joyson Galbao, Seung-Cheol Lee, Satadeep Bhattacharjee, Dominik Eder, Alexey Cherevan, and Dharmapura H. K. Murthy
ACS Applied Materials & Interfaces
Abstract
Unveiling Valence State-Dependent Photocatalytic Water Splitting Activity and Photocathodic Behavior in Visible Light-Active Iridium-Doped BaTiO3
Despite having favorable energetics and tunable optoelectronic properties, utilization of BaTiO3 (BTO) for photocatalytic reactions is limited by its absorption only in the ultraviolet region. To address this challenge, BTO is doped with iridium (Ir) to induce visible light absorption. The visible light-induced photocatalytic H2 generation efficiency is enhanced by 2 orders of magnitude on selective conversion of the Ir valence state from Ir 4+ to Ir 3+. To understand such intriguing behavior, valence state-dependent changes in the optoelectronic, structural, and surface properties and electronic band structure are comprehensively investigated. The effect of electron occupancy change between Ir 4+ (t 2g 5 e g 0) and Ir 3+ (t 2g 6 e g 0) and their energetic positions within the band gap is found to significantly influence H 2 generation. Besides this, converting Ir 4+ to Ir 3+ enhanced the photocathodic current and lowered the onset potential. Results aid in designing photocatalysts to efficiently use low-energy photons for enhancing solar H2 production in these emerging BTO-based photocatalysts. Collectively, the observations made in this work highlight the promising application of Ir 3+ :BTO in z-scheme photocatalysis.
90
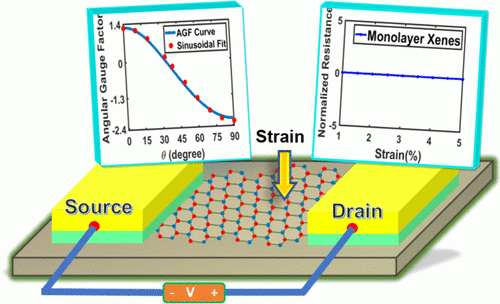
Density Functional Theory of Straintronics Using the Monolayer-Xene Platform: A Comparative Study
Swastik Sahoo, Namitha Anna Koshi, Seung-Cheol Lee, Satadeep Bhattacharjee, and Bhaskaran Muralidharan
ACS Applied Nano Materials
Abstract
Density Functional Theory of Straintronics Using the Monolayer-Xene Platform: A Comparative Study
Monolayer silicene is a front runner in the two-dimensional (2D)-Xene family, which also comprises germanene, stanene, and phosphorene, to name a few, due to its compatibility with current silicon fabrication technology. Here, we investigate the utility of 2D-Xenes for straintronics using the ab initio density functional theory (DFT) coupled with quantum transport based on the Landauer formalism. With a rigorous band structure analysis, we show the effect of strain on the K-point and calculate the directional piezoresistances for the buckled Xenes as per their critical strain limit. Further, we compare the relevant gauge factors (GFs) and their sinusoidal dependencies on the transport angle akin to those of silicene and graphene. The strain-insensitive transport angles corresponding to the zero gauge factors for silicene and germanene are 81 and 34° for armchair (AC) and zigzag (ZZ) strains, respectively. As the strain limit is increased to 10% in stanene, there are notable changes in the fundamental parameters, which entail a change in the critical angle along the armchair (69°) and zigzag (34°) directions. The small values of gauge factors can be attributed to their stable Dirac cones and strain-independent valley degeneracies. We also explore conductance modulation, which is quantized in nature and exhibits a variation pattern similar to that of other transport parameters against applied strain. Based on the obtained results, we propose the buckled Xenes as an interconnect in flexible electronics and that they are promising candidates for various applications in straintronics.
89
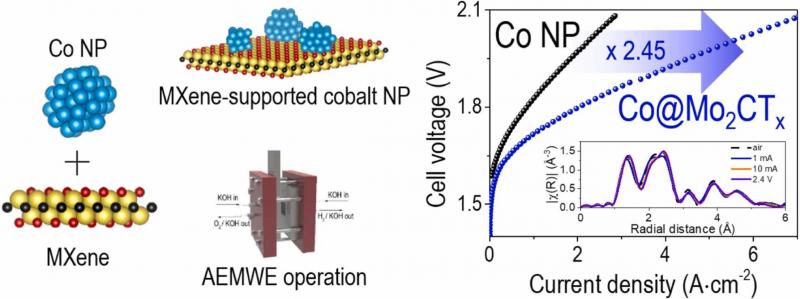
Unveiling the role of catalytically active MXene supports in enhancing the performance and durability of cobalt oxygen evolution reaction catalysts for anion exchange membrane water electrolyzers
Young Sang Park, Ari Chae, Gwan Hyun Choi, Swetarekha Ram, Seung-Cheol Lee, Satadeep Bhattacharjee, Jiyoon Jung, Hyo Sang Jeon, Cheol-Hee Ahn, Seung Sang Hwang, Dong-Yeun Koh, Insik In, Taegon Oh, Seon Joon Kim, Chong Min Koo, Albert S. Lee
Applied Catalysis B: Environment and Energy
Abstract
Unveiling the role of catalytically active MXene supports in enhancing the performance and durability of cobalt oxygen evolution reaction catalysts for anion exchange membrane water electrolyzers
The role of 2D transition metal carbides, also known as MXenes, as active catalyst supports in Co-based oxygen evolution reaction (OER) catalysts was elucidated through a combination of experimental and computation electrochemistry. Through facile seeding of commericial Co nanoparticles on three different MXene supports (Ti3C2Tx, Mo2Ti2C3Tx, Mo2CTx), Co@MXene catalysts were prepared and their electrochemical properties examined for alkaline OER electrocatalysts. The OER activity enhancement of Co was significantly improved for Mo2CTx and Mo2Ti2C3Tx supports, but marginal on the Ti3C2Tx in rotating disk electrode and membrane electrode assembly tests. The Co@Mo2CTx exhibited the highest anion exchange water electrolysis performance of 2.11 A cm−2 at 1.8 V with over 700 h of stable performance, exceeding previous benchmarks for non-platinum group (non-PGM) metal OER catalysts. The superior performance was attributed to the strong chemical interaction of Co nanoparticle with the Mo2CTx MXene support. Insights into the electrochemical and chemical oxidation according to MXene type as related to cell durability, as well the effect of electrical conductivity and inherent boosting of electrocatalytic activity of Mo-based MXenes elucidated through density functional theory (DFT) calculations helped explain the performance and durability enhancement of Mo-based MXene supports over Ti3C2Tx supports.
88
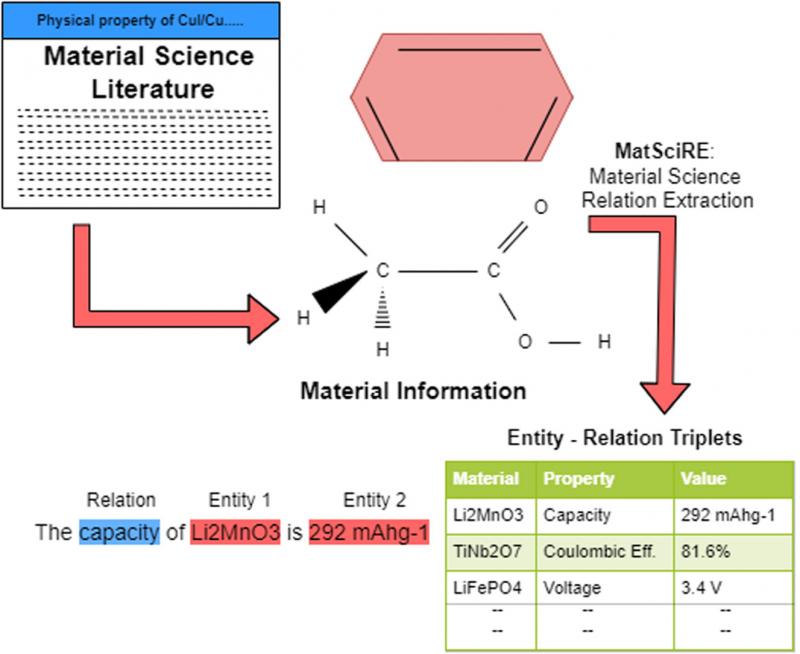
MatSciRE: Leveraging pointer networks to automate entity and relation extraction for material science knowledge-base construction
Ankan Mullick, Akash Ghosh, G. Sai Chaitanya, Samir Ghui, Tapas Nayak, Seung-Cheol Lee, Satadeep Bhattacharjee, Pawan Goyal
Computational Materials Science
Abstract
MatSciRE: Leveraging pointer networks to automate entity and relation extraction for material science knowledge-base construction
Material science literature is a rich source of factual information about various categories of entities (like materials and compositions) and various relations between these entities, such as conductivity, voltage, etc. Automatically extracting this information to generate a material science knowledge base is a challenging task. In this paper, we propose MatSciRE (Material Science Relation Extractor), a Pointer Network-based encoder–decoder framework, to jointly extract entities and relations from material science articles as a triplet (entity1, relation, entity2). Specifically, we target the battery materials and identify five relations to work on — conductivity, coulombic efficiency, capacity, voltage, and energy. Our proposed approach achieved a much better F1-score (0.771) than a previous attempt using ChemDataExtractor (0.716). The overall graphical framework of MatSciRE is shown in Fig. 1. The material information is extracted from material science literature in the form of entity–relation triplets using MatSciRE.
87
Instant Self-Assembly of Functionalized MXenes in Organic Solvents: General Fabrication to High-Performance Chemical Gas Sensors
Soobin Kim, Tae Yun Ko, Ajit K. Jena, Arun S. Nissimagoudar, Juyun Lee, Seongeun Lee, Taegon Oh, Yun Chan Kang, Insik In, Satadeep Bhattacharjee, Chong Min Koo, Seung-Cheol Lee, Seon Joon Kim
Advanced Functional Materials
Abstract
Instant Self-Assembly of Functionalized MXenes in Organic Solvents: General Fabrication to High-Performance Chemical Gas Sensors
MXenes are a promising class of two-dimensional transition metal carbides, nitrides, and carbonitrides, widely utilized in diverse fields such as energy storage, electromagnetic shielding, electrocatalysis, and sensing applications. Their potential in chemical sensing is particularly noteworthy, where optimizing surface chemistry for strong interaction with target analytes and increasing surface area for efficient gas adsorption are crucial factors. In this study, a versatile and general self-assembly method for fabricating nanometer-scale thin films of surface-functionalized MXene, enabling high-performance gas sensors is developed. By dropping MXene dispersed in organic solvents onto nonsolvents, rapid formation of nanometer-scale films is achieved. This method allows easy adjustment of film properties by using different solvent-nonsolvent combinations, leading to improved optoelectronic properties compared to conventional techniques. The surface-functionalized MXenes using ADOPA ligands greatly enhance the gas response and long-term environmental stability compared to pristine MXenes. Computational methods are also employed to gain insights into the molecular interactions and changes in electronic structure that contribute to the enhanced sensing properties. Furthermore, the environmental stability of MXene sensors is largely enhanced after surface functionalization, which can be attributed to increased surface hydrophobicity. Overall, this innovative technique opens up opportunities for tailoring MXene thin films for specific applications.
86
Revealing the origin of the topological Hall effect in the centrosymmetric shape memory Heusler alloy Mn 2 NiGa : A combined experimental and theoretical investigation
Shivani Rastogi, Nisha Shahi, Vishal Kumar, Gaurav K. Shukla, Satadeep Bhattacharjee, and Sanjay Singh
Physical Review B
Abstract
Revealing the origin of the topological Hall effect in the centrosymmetric shape memory Heusler alloy Mn 2 NiGa : A combined experimental and theoretical investigation
Skyrmions are localized swirling noncoplanar spin textures offering a promising revolution in future spintronic applications. These topologically nontrivial spin textures lead to an additional contribution to the Hall effect, called the topological Hall effect. Here, we investigate the origin of the topological Hall effect—a trademark of skyrmions—in a centrosymmetric shape memory Heusler alloy (SMHA) Mn2NiGa. The magnetization measurement unveils the presence of austenite to martensite transition in the studied system. The topological Hall effect (THE) in the present system is examined experimentally and theoretically. The presence of a large THE in the austenite (cubic) phase of the system strongly suggests that the observed THE in Mn2NiGa cannot be attributed to the antiskyrmions stabilized by D2d symmetry as reported earlier. To comprehend the underlying mechanism behind the origin of THE, we have performed micromagnetic simulations for a range of magnetic field with a small value of DMI (local DMI) to consider the possible impact of earlier reported atomic disorder in the centrosymmetric SMHA Mn2NiGa. The results showed the stabilization of Néel-type skyrmions, which can be assigned to the expected local symmetry breaking at the interface of disorder originated ferromagnetic nanoclusters and ferrimagnetic lattice of the system. A theoretical calculation of topological Hall resistivity by utilizing micromagnetic simulations is performed, which is of the same order as the experimentally obtained values in the both martensite and austenite phases.